PDF Publication Title:
Text from PDF Page: 005
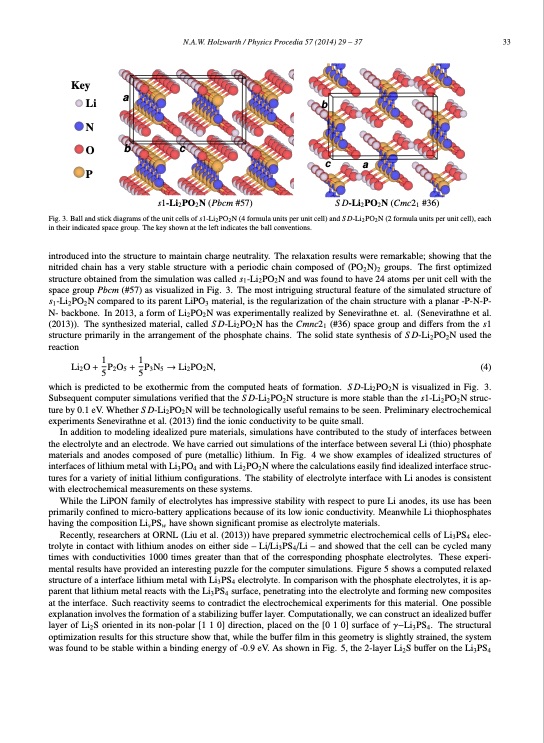
N.A.W. Holzwarth / Physics Procedia 57 (2014) 29 – 37 33 a bc Key Li b N O P c a s1-Li2PO2N (Pbcm #57) Fig. 3. Ball and stick diagrams of the unit cells of s1-Li2 PO2 N (4 formula units per unit cell) and S D-Li2 PO2 N (2 formula units per unit cell), each in their indicated space group. The key shown at the left indicates the ball conventions. introduced into the structure to maintain charge neutrality. The relaxation results were remarkable; showing that the nitrided chain has a very stable structure with a periodic chain composed of (PO2N)2 groups. The first optimized structure obtained from the simulation was called s1-Li2PO2N and was found to have 24 atoms per unit cell with the space group Pbcm (#57) as visualized in Fig. 3. The most intriguing structural feature of the simulated structure of s1-Li2PO2N compared to its parent LiPO3 material, is the regularization of the chain structure with a planar -P-N-P- N- backbone. In 2013, a form of Li2PO2N was experimentally realized by Senevirathne et. al. (Senevirathne et al. (2013)). The synthesized material, called S D-Li2PO2N has the Cmnc21 (#36) space group and differs from the s1 structure primarily in the arrangement of the phosphate chains. The solid state synthesis of S D-Li2PO2N used the reaction Li2O + 15P2O5 + 15P3N5 → Li2PO2N, (4) which is predicted to be exothermic from the computed heats of formation. S D-Li2PO2N is visualized in Fig. 3. Subsequent computer simulations verified that the S D-Li2PO2N structure is more stable than the s1-Li2PO2N struc- ture by 0.1 eV. Whether S D-Li2PO2N will be technologically useful remains to be seen. Preliminary electrochemical experiments Senevirathne et al. (2013) find the ionic conductivity to be quite small. In addition to modeling idealized pure materials, simulations have contributed to the study of interfaces between the electrolyte and an electrode. We have carried out simulations of the interface between several Li (thio) phosphate materials and anodes composed of pure (metallic) lithium. In Fig. 4 we show examples of idealized structures of interfaces of lithium metal with Li3PO4 and with Li2PO2N where the calculations easily find idealized interface struc- tures for a variety of initial lithium configurations. The stability of electrolyte interface with Li anodes is consistent with electrochemical measurements on these systems. While the LiPON family of electrolytes has impressive stability with respect to pure Li anodes, its use has been primarily confined to micro-battery applications because of its low ionic conductivity. Meanwhile Li thiophosphates having the composition LivPSw have shown significant promise as electrolyte materials. Recently, researchers at ORNL (Liu et al. (2013)) have prepared symmetric electrochemical cells of Li3PS4 elec- trolyte in contact with lithium anodes on either side – Li/Li3PS4/Li – and showed that the cell can be cycled many times with conductivities 1000 times greater than that of the corresponding phosphate electrolytes. These experi- mental results have provided an interesting puzzle for the computer simulations. Figure 5 shows a computed relaxed structure of a interface lithium metal with Li3PS4 electrolyte. In comparison with the phosphate electrolytes, it is ap- parent that lithium metal reacts with the Li3PS4 surface, penetrating into the electrolyte and forming new composites at the interface. Such reactivity seems to contradict the electrochemical experiments for this material. One possible explanation involves the formation of a stabilizing buffer layer. Computationally, we can construct an idealized buffer layer of Li2S oriented in its non-polar [1 1 0] direction, placed on the [0 1 0] surface of γ−Li3PS4. The structural optimization results for this structure show that, while the buffer film in this geometry is slightly strained, the system was found to be stable within a binding energy of -0.9 eV. As shown in Fig. 5, the 2-layer Li2S buffer on the Li3PS4 S D-Li2PO2N (Cmc21 #36)PDF Image | First Principles Modeling of Electrolyte Materials in All-Solid-State Batteries
PDF Search Title:
First Principles Modeling of Electrolyte Materials in All-Solid-State BatteriesOriginal File Name Searched:
1-s2-0-S1875389214002727-main.pdfDIY PDF Search: Google It | Yahoo | Bing
Sulfur Deposition on Carbon Nanofibers using Supercritical CO2 Sulfur Deposition on Carbon Nanofibers using Supercritical CO2. Gamma sulfur also known as mother of pearl sulfur and nacreous sulfur... More Info
CO2 Organic Rankine Cycle Experimenter Platform The supercritical CO2 phase change system is both a heat pump and organic rankine cycle which can be used for those purposes and as a supercritical extractor for advanced subcritical and supercritical extraction technology. Uses include producing nanoparticles, precious metal CO2 extraction, lithium battery recycling, and other applications... More Info
| CONTACT TEL: 608-238-6001 Email: greg@infinityturbine.com | RSS | AMP |